|
第二节 过渡态理论
反应速度是决定反应历程的动力学因素,一般有机反应,在少数情况下,才能建立平衡。又因为有机反应比较复杂,在一个反应中往往有其他副反应的竞争。实际上许多生产中应用的有机反应也并不是按达到平衡条件,而往往只是按竞争反应中速度最快的反应来设计的。因此,有机反应的进程大多决定于动力学因素。为了能动地控制反应速度,有效地进行有机合成和工业生产,就要研究和了解反应速度理论问题。在目前,关于反应速度的理论有两种,一种是碰撞理论,另一种是过渡态理论。前者是在气体分子运动论的基础上建立起来的,后者是在统计力学和量子力学的发展中形成的。我们仅讨论过渡态理论.
过渡态理论也称活化络合物理论,它的主要内容建立在某些基本假定基础之上。最基本的假定是:反应物分子在互相接近的过程中先被活化形成活化络合物即过渡态,过渡态再以一定的速度分解为产物,过渡态是反应物与产物之间的中间状态。
反应物→过渡态→产物
过渡态(transition
state
TS)在反应进程中位于能量最高处,如反应进程图所示,反应进程是指从反应物到产物所经过的能量要求最低的途径。在反应坐标上位能起伏的曲线,叫做反应位能曲线。例如:从A+B-C到A-B+C的反应可有两条途径,一条是B―C之间的键先断裂,生成的B再与A结合变成A-B,另一条途径是A从背面接近B,A-B键的生成与B―C键的断裂协同进行,成键所放出的能量,一部分可提供旧键断裂的需要,要形成过渡态所需要的能量一般要比旧键完全断裂(离解成自由基或离子)所需的能量低。因此一条途径,即经过过渡态的反应途径从能量上来讲是有利的。
A +
B-C →A-B + C
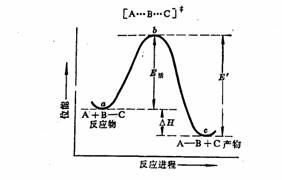
由稳定的反应物分子过渡到活化络合物的过程叫做活化过程,活化过程所吸收的能量称为活化能,因此,活化能是基态反应物平均能量与过渡态之间的能量差。凡有共价
键断裂的反应都需要活化能。在同一温度下,活化能低的反应,其速度较快。
如果反应是可逆的,则正逆反应(在同一条件下进行)必然是以同一机理而进行,这叫做微观可逆性原理。正反应的活化能减去逆反应的活化能等于反应的热效应。
E―E′=△H
吸热反应的活化能比△H大。这里要注意活化能(E)和反应热(△H)之间是不存在直接关系的,不能从△H来预测E的大小。反应热是产物与反应物的焓差,在一般情况下,近似等于产物与反应物的内能差,所以它可以从反应中断裂和形成化学键的能量变化近似地计算出来。而活化能则是过渡态与反应物的内能差,除了少数反应的活化能可以从理论上估算外,一般只能通过温度和反应速度的关系由实验测得。决定反应速度的是活化能E,是能垒高度,而不是两个能谷的高度差△H。例如,某些催化反应,催化剂的作用只是降低活化能,加速反应速度,并不改变热效应△H的数值。即使是放热反应也需要一定的活化能。不能错误地认为,吸热反应的活化能E一定就大,而放热反应的活化能E必然就小。
过渡态理论的另一个假定是过渡态的空间结构与能量相近的分子(反应物或产物)类似。例如,在放热反应中过渡态的能量与反应物更接近,它的结构也应该与反应物近似;在吸热反应中,过渡态的能量和结构则与产物近似。由于过渡态在反应途径中处于能量最高点,很不稳定,其寿命接近于零,不能用实验方法来观察,所以只能根据结构相近则内能相近的原则,反过来对它的结构作一些理论上的推测或假设。
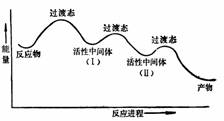
以上所讨论的都是对基元反应而言,而许多反应是分步进行的,在这些多步反应中会存在几个过渡态,两个过渡态之间的能谷是反应的活性中间体。活化能大的步骤速度慢,是决定反应速度的步骤。如下图:
共1页
上一节
下一节
|